ALS: What Is Lou Gehrig’s Disease?
Officially, it's amyotrophic lateral sclerosis, or ALS, but it's more commonly known as Lou Gehrig's disease. Here's what you need to know about this devastating condition.
ALS—the abbreviation for amyotrophic lateral sclerosis—is a rare neurological disorder that’s eventually fatal. It was July 4, 1939, when Lou Gehrig made his famous “luckiest man alive” speech at Yankee Stadium, an event that helped give widespread attention to ALS. Gehrig, when he was diagnosed, was only 36 years old and in the 17th year of his Hall of Fame career. After diagnosis, the great New York Yankees slugger went quickly, succumbing in 1941 before turning 38. His death from ALS gave the affliction a more commonly known name: Lou Gehrig’s disease.
More recently, in 2014, the disease received an important wave of attention because of the ALS Association‘s “Ice Bucket Challenge,” which involved people voluntarily getting doused by freezing water as part of an awareness campaign and fund-raising drive. It raised more than $115 for research.

Professor Stephen Hawking is pictured here in 2006 during a conference in Italy.
And in March 2018, physicist Stephen Hawking died of ALS, though he was 76 and had been diagnosed at the age of 21. (Click here for Scientific American‘s feature explaining how Hawking beat the odds and survived so long with ALS.)
So what is amyotrophic lateral sclerosis? Below, we explore the symptoms, causes, and diagnosis of ALS. We also touch on the treatment of the disease.
ALS: The Details
According to the National Institute for Neurological Disorders and Stroke (NINDS), “Amyotrophic lateral sclerosis (ALS) is a group of rare neurological diseases that mainly involve the nerve cells (neurons) responsible for controlling voluntary muscle movement.” This results in degeneration of nerves in the brain and spinal cord, and progressive loss of the ability to chew, swallow, walk, and talk.
The disease is progressive, meaning symptoms get worse over time, until it is ultimately fatal. There is currently no cure.
Fortunately, ALS is rare. The ALS Association reports that there are around 6,400 new cases in the United States every year, with approximately 20,000 people living with the condition. The incidence is on the rise, perhaps because we are all living longer and because of an increase in environmental triggers.
What Causes ALS?
ALS is the most common form of a group of diseases known as motor neuron diseases (MNDs). In fact, it’s referred to as “classical motor neuron disease.” Most cases of ALS are sporadic, with no family history; approximately 5 to 10 percent are inherited or familial.
Sporadic ALS: Researchers have found clues as to the potential triggers for ALS. They may include environmental factors such as smoking, toxins (including lead and diesel fumes), infections, trauma, poor diet, overexercising, and behavioral and occupational factors.
The mechanism for nerve degeneration is thought to involve oxidation, autoimmune reactions, neurochemical imbalances, and abnormal protein build. Veterans and athletes are at higher risk, and the risk for men is slightly higher than for women.
Familial ALS: Researchers have found more than a dozen genetic mutations—including the SOD1 and C9ORF72 genes—that are linked to familial ALS. According to the NIH Genetics Home Reference, most cases are “autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.”
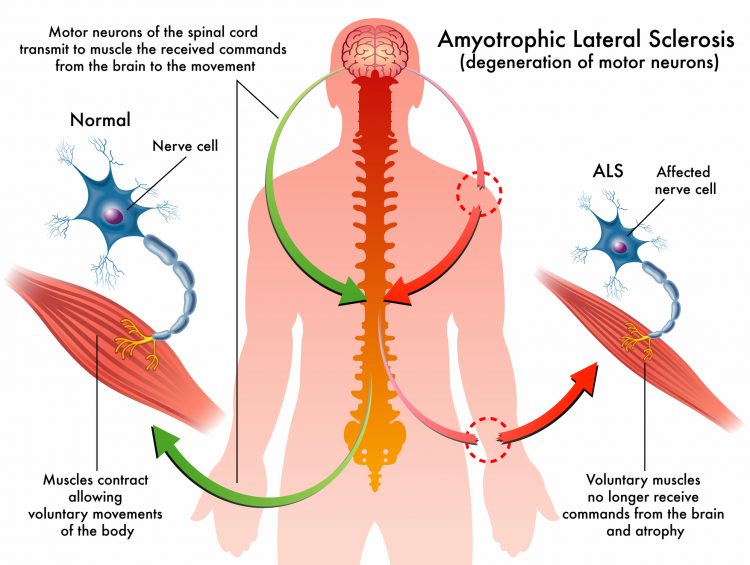
The graphic here illustrates the impact of amyotrophic lateral sclerosis (ALS).
What Are the Symptoms of ALS?
ALS most commonly develops between the ages of 55 and 75. It usually begins with subtle, vague symptoms. Early symptoms, according to the NINDS, can include:
- Muscle twitches in the arm, leg, shoulder, or tongue
- Muscle cramps
- Tight and stiff muscles (spasticity)
- Slurred and nasal speech (“bulbar” symptoms)
- Difficulty chewing or swallowing
- Muscle weakness affecting an arm, a leg, neck, or diaphragm, which may present as stumbling when walking or having difficulty buttoning a shirt.
Progression of ALS is variable but involves a gradual increase in muscle weakness (often one-sided), pain and wasting (atrophy), problems with moving, swallowing (dysphagia), speaking (dysarthria), and breathing.
Mobility and completion of simple tasks becomes increasingly difficult as the disease advances. Choking may occur due to difficulty swallowing. Weight loss also occurs due to increased metabolism, and malnutrition is common.
Sufferers eventually become wheelchair-bound and dependant on others for activities of daily living. Degeneration of the respiratory nerves leads to weakening of respiratory muscles, recurrent pneumonias, and eventually the need for artificial ventilation.
Cognitive abilities are often spared early on in ALS (in 50 to 80 percent of sufferers), which is a blessing and a curse, as many sufferers go on to develop depression. However, thinking speed and attention may be impacted, and emotional lability (varying extremes of emotion) may occur. Dementia can occur later as the condition progresses.
Diagnosing ALS
If you have any of the symptoms mentioned above, go to see your doctor. He or she will take a medical and family history and perform a physical examination and may organize some basic tests, or refer you to a neurologist.
Your doctor or neurologist will want to exclude other causes of your symptoms. There is no conclusive test for ALS and it may take months to reach a diagnosis.
Investigations may include:
- Blood tests can identify (and rule out) CBC, magnesium, kidney function, electrolytes, thyroid function, blood chemistry, and viruses including HIV and polio.
- Genetic testing may also be useful.
- Electromyography (EMG) is a technique for measuring the electrical activity in muscles fibers.
- Nerve conduction studies (NCS) measure electrical activity in nerves and muscles.
- Magnetic resonance imaging (MRI) scans provide detailed 3D imaging of the brain and spinal cord. This is helpful in excluding problems such as tumors and abnormalities of the spine.
- Multiple positron emission tomography (PET) imaging is being used in some research studies.
Treatment of ALS
There is no cure for ALS. Treatment focuses on managing symptoms, preventing complications, and reducing disability. The average cost of care has been estimated at $50,000 per patient per year, and a coordinated multidisciplinary team approach is needed.
- MEDICAL TEAM
- Several specialists may be involved with care: neurologist, physical therapist, occupational therapist, physiatrist, nutritionist, gastroenterologist, respiratory therapist, speech therapist, nurses, and palliative care practitioners. A social worker, psychiatrist, and psychotherapist may also be involved.
- MEDICATIONS
- Disease modifying: Riluzole (Rilutek) and edaravone (Radicava) are FDA-approved drugs for ALS. They may slow progression of the disease and prolong life for up to several months.
- Muscle symptoms: Baclofen, quinine bisulphate, and diazepam may give some relief.
- Pain management: nonsteroidal anti-inflammatory drugs (NSAIDs), anticonvulsants, tricyclic antidepressants, and opiates.
- Emotional problems: Neurodex, a vitamins B-1, B-6, and B-12.
Research is ongoing into the potential benefits of coenzyme Q10, COX-2 inhibitors, creatine and minocycline.
Prognosis for ALS
After diagnosis, most people with ALS live between two and five years. Twenty percent of sufferers survive over five years, while 10 percent survive over 10 years. Respiratory failure is the most common cause of death.
 Vestibular Migraine Diet
Vestibular Migraine Diet  Bone Spurs on the Spine
Bone Spurs on the Spine  Knee Injections for Osteoarthritis Pain Relief
Knee Injections for Osteoarthritis Pain Relief 
On July 4, 1939, Lou Gehrig made his "luckiest man alive" speech at Yankee Stadium. Just two years later, he died from ALS complications. The impact of his death at such a young age—he was 37—prompted people to begin referring to ALS as "Lou Gehrig's disease."