Cystic Fibrosis: Understanding a Genetic Respiratory Disease
What is cystic fibrosis? It's a genetic disease that ravages the lungs and other organs in the body. Let’s explore this complex inherited disease, addressing its causes, symptoms, and treatment.
Cystic fibrosis is a genetic or inherited disease of the mucus and sweat glands. It affects approximately 30,000 Americans, leading to chronic health problems and premature death. Advances in treatment, however, have dramatically improved outcomes in the past 20 years.
In people with cystic fibrosis, or CF, the mucus produced by the organs of the body are thick and sticky. CF mainly affects the lungs, digestive tract, pancreas, liver, sinuses, and sex organs. The lungs become prone to breathing problems, severe infections, and progressive damage. Digestion is also frequently affected, leading to malabsorption of nutrients.
“CF also causes your sweat to become very salty,” according to the National Heart, Lung, and Blood Institute. “Thus, when you sweat, you lose large amounts of salt. This can upset the balance of minerals in your blood and cause many health problems. Examples of these problems include dehydration (a lack of fluid in your body), increased heart rate, fatigue (tiredness), weakness, decreased blood pressure, heat stroke, and, rarely, death.”
KEY FACTS ABOUT CYSTIC FIBROSIS
- 1 in 2,500 babies are born with cystic fibrosis.
- Cystic fibrosis is an autosomal recessive genetic disease.
- CF affects the lungs, digestive, and reproductive tract.
- More than 75 percent of people with CF are diagnosed by age 2.
- Life expectancy is improving year on year, due to medical advances.
Cystic Fibrosis: The Disease’s Process
Experts at the American Lung Association explain that “people with CF don’t make enough or make an abnormal version of a protein called cystic fibrosis transmembrane regulator (CFTR). CFTR is present on the cell surface in many organs and regulates the movement of salt-sodium (Na) and chloride (Cl) ions, as well as water across the cell surface.”
Cystic fibrosis is a complex disease and affects people in different ways, including these areas:
- Respiratory tract. With cystic fibrosis, the lungs produce thick, sticky mucus, which builds up in the airways and allows infections to multiply and cause disease (bronchitis, pneumonia, pneumothorax, bronchiectasis). Nasal polyps and sinus infections also are very common. Suffers may become ill with infections that don’t normally affect other people; among these infections are Pseudomonas (bacteria) and Aspergillus (fungus/ black mold). Respiratory failure is the most common cause of death.
- Pancreas. Mucus also builds up in the pancreatic ducts leading to scarring and a reduction in secretion of pancreatic enzymes, essential for digestion. Fat and nutrient absorption is reduced leading to malnutrition and poor growth. It can also lead to “cystic fibrosis-related diabetes.”
- Liver. Mucus blocks the bile ducts causing liver damage, cirrhosis, and malabsorption.
- Digestive tract. Secretions can be thicker sometimes causing malabsorption, bowel problems and even intestinal blockages.
- Reproductive tract. Infertility occurs in most men and decreased fertility in most women.
In the 1950s, many CF sufferers would die in childhood, but medical breakthroughs have improved prognosis considerably; the average survival age is close to 40. Most are able to live fulfilling lives. Individuals, however, need intensive daily treatment and may require frequent hospital admissions.
What Causes Cystic Fibrosis?
One in 2,500 babies are born every year with cystic fibrosis. It’s an autosomal recessive genetic disease, meaning that the individual must have two copies of the defective CFTR gene: one copy from each parent. (CFTR stands for “cystic fibrosis transmembrane regulator.”) According to estimates, 1 in 20 Americans are carriers, but they may be unaware unless they have a child diagnosed with CF.
Each offspring of two CF carrier parents has a 25 percent chance of developing cystic fibrosis and a 50 percent chance of becoming a carrier.
There are more than 1,700 known mutations (slight abnormalities) of the gene. Genetic screening will pick up the common types, but miss some of the rarer ones.
All racial groups are affected, but CF is most common in Caucasians.
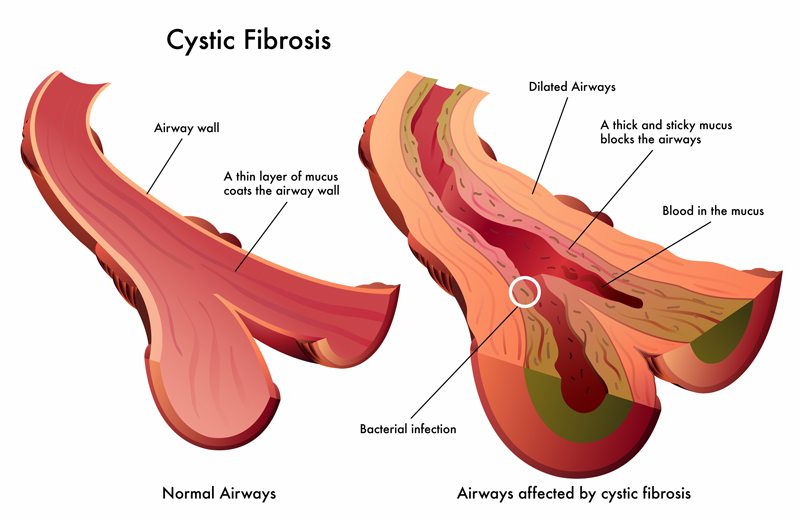
Cystic fibrosis is marked by airways in the lungs that become blocked by a thick, sticky mucus.
Symptoms of Cystic Fibrosis
The presentation and course of CF is extremely variable. Cystic fibrosis can present soon after birth or, in mild cases, as late as the teenage years. When it presents in infancy early signs may include salty tasting skin, respiratory distress, failure to pass the first stool, severe constipation, and failure to thrive (not growing and gaining weight).
According to the Cystic Fibrosis Foundation, common symptoms include:
- Very salty-tasting skin
- Persistent coughing, at times with phlegm (and blood)
- Frequent lung infections including pneumonia or bronchitis
- Wheezing or shortness of breath
- Poor growth or weight gain in spite of a good appetite
- Frequent greasy, bulky stools or difficulty with bowel movements
- Infertility
Additional chronic problems include:
- Dehydration, fatigue, low blood pressure, and heat intolerance
- Pancreatitis
- Rectal prolapse
- Liver disease
- Osteoporosis
- Diabetes
- Gallstones
Diagnosing Cystic Fibrosis
There are multiple ways that the medical community tests for cystic fibrosis:
- Newborn screening. In the U.S., all newborns are screened for the faulty CFTR genes.
- Sweat test. If a doctor suspects CF they will order a “sweat test” which measures the amount of salt in sweat.
- Genetic tests. Will determine the presence of a CFTR defect.
- Chest x-ray. Is used to monitor progression of the disease and complications.
- Lung function tests. These tests are a performed regularly on people with CF.
- A sputum culture. A sputum sample will be taken at routine follow up and if any respiratory symptoms develop in order to detect and guide treatment infections.
- Prenatal genetic screening. These tests may be offered to families with a history of CF and are done as part of an amniocentesis and chorionic villus sampling, following genetic counseling.
- Cystic fibrosis carrier testing. Those with a family history of CF may opt for genetic counselling and screening before considering having a baby.
Treatments for Cystic Fibrosis
Treatment plans need to be highly personalized, depending on issues that arise. Most patients are treated at one of more than 120 specialized care centers. The team of specialists will include pediatric, respiratory, and gastroenterology physicians as well as dietitians, social workers, respiratory and physical therapists, and CF nurses.
Daily therapy for cystic fibrosis may include the following.
- Airway clearance techniques (a.k.a. chest physiotherapy or pulmonary rehabilitation) are needed daily to help reduce mucus and the risk of infection. These procedures can be done by the individual (when older), family members, friends, or respiratory therapists. Vibrating inflatable vests may also help.
- Inhaled medication (via inhaler or nebulizer) can help open the airways or thin the mucus.
- Pancreatic enzyme supplement capsules are taken with most meals, to improve nutrient absorption.
- Nutritional therapy may include vitamin supplements, high-calorie shakes, and a high-salt diet. Some individuals require a feeding tube.
SOURCES & RESOURCES
Additional treatments may include the following.
- CFTR modulators are a new group of drugs (the first came out in 2012) that treat the root cause of cystic fibrosis. It is hoped that they will improve prognosis by several years, if not decades.
- Oxygen therapy may be needed during bouts of infection or deterioration in lung function.
- Lung transplant may be considered for those with severe lung disease.
CF is a very active area of research. The goal is to ultimately find a way to extend lifespan and perhaps to cure this devastating disease. Biotechnology and stem cell therapies may hold the key.
Living with Cystic Fibrosis
In addition to medical care and daily therapy, there are many things that an individual and his or her family can do to improve well-being when living with CF. Strategies include:
- Regular exercise. Under medical supervision, it can help improve lung function and overall well-being.
- A healthy, balanced diet under the supervision of a dietitian or nutritionist. Good hydration is also essential.
- Avoid sick people when possible.
- Good hand-hygiene will reduce the risk of infection.
- Keep your home free of black mold, which may be due to the mold aspergillus.
- Psychological counseling and/or group support will help individuals and their families cope with the emotional side of living with this chronic illness.
- Avoid smoking and tobacco smoke.
 What is a Pulse Oximeter?
What is a Pulse Oximeter?  Lung Detox or Lung Cleanse: Can You Clean Out Your Lungs Naturally?
Lung Detox or Lung Cleanse: Can You Clean Out Your Lungs Naturally?  Are There House Plants That Clean the Air?
Are There House Plants That Clean the Air? 
Cystic fibrosis can result in a lung infection caused by the bacteria Pseudomonas aeruginosa.
Illustration 124562204 © Katerynakon - Dreamstime.com